What is Phylogenetics?
Phylogenetics is the study of linkages between different species. These linkages refer to similar DNA sequences for similarly functioning proteins. These relationships are established and displayed through phylogenetic trees. Trees are a way to visualize and physically connect separate taxa together and determine similarities. Species that are similar will be grouped together, while species that are less related are placed further away or put as outliers. There are many different strategies to creating a phylogenetic tree. They include Neighbor Joining, Minimum Parsimony, Maximum Likelihood and the Bayesian Methods. Depending on what type of research you are conducting, what information you have as well as time, particular methods for constructing a tree could be more beneficial. They can also be used to infer synonymous and non-synonymous mutations, how many mutations are predicted per site and how the accumulation of such mutations over time have influenced divergence of species.
Phylogenetics is the study of linkages between different species. These linkages refer to similar DNA sequences for similarly functioning proteins. These relationships are established and displayed through phylogenetic trees. Trees are a way to visualize and physically connect separate taxa together and determine similarities. Species that are similar will be grouped together, while species that are less related are placed further away or put as outliers. There are many different strategies to creating a phylogenetic tree. They include Neighbor Joining, Minimum Parsimony, Maximum Likelihood and the Bayesian Methods. Depending on what type of research you are conducting, what information you have as well as time, particular methods for constructing a tree could be more beneficial. They can also be used to infer synonymous and non-synonymous mutations, how many mutations are predicted per site and how the accumulation of such mutations over time have influenced divergence of species.
|
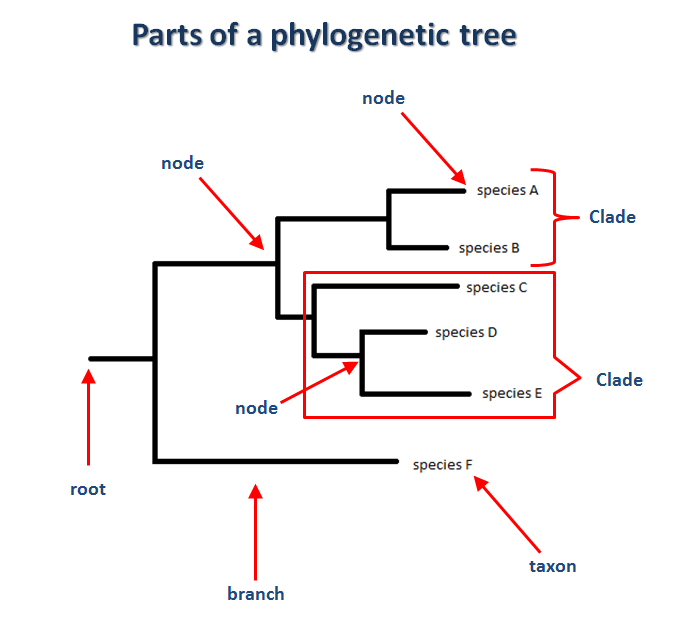
Phylogenetic trees all have the same basic features that make all trees easy to interpret. To start, trees can be rooted or unrooted. When a tree is rooted it simply means that the species share a common known ancestor that is based on time. A node is a break point in which a species diverged from it's ancestor. A branch represents a unique lineage that has persisted over time. Usually the length indicates how much time has passed between the divergence and the species that we see in present day. A clade a group of species that share the same ancestor or a common node.
|
Phylogenetic Tree Construction
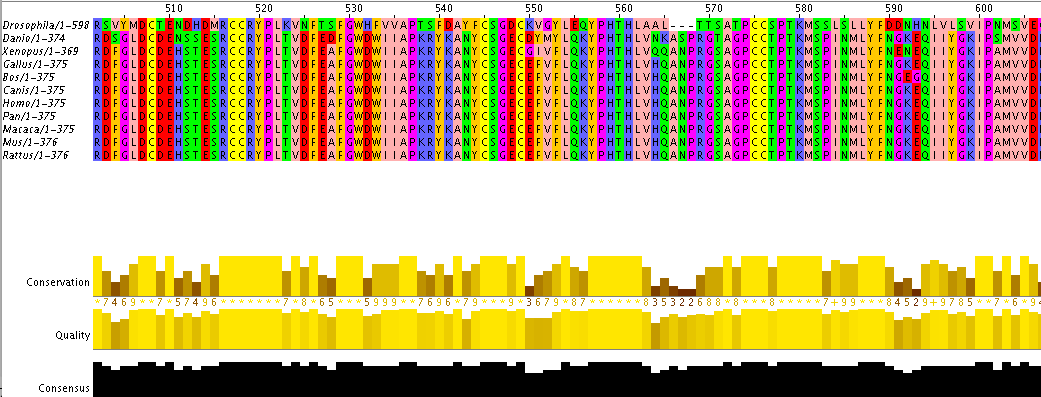
Phylogenetic trees are constructed by comparing DNA sequences determined through next-generation sequencing. Sequences are then aligned using ClustalW in a way that allows for the same proteins and domains to be aligned between genes of different species. The following image is an example of how sequences from homologs of the MSTN gene look when aligned. Each amino acid is assigned a letter as well as a color, making it easy to interpret quickly. Based on the alignment, different algorithms are used in order to determine the most similar sequences set of sequences and thus should be placed together on a phylogenetic tree. Different algorithms are used to asses different sets of data and are better at handling different information such as gaps, highly divergent sequences and even large amounts of data. These different techniques are described further below.
Minimum Evolution
Minimum Evolution Phylogenetic Trees utilize a distance method to determine which species are most closely related. This means that the shortest trees are most likely to be the most correct. This technique uses distances matrixes to compare the distance between all the sequences and then use this information to construct the shortest tree. They are very computationally efficient and are used when analyzing large amounts of data with a low level of divergence between all the sequences.
Neighbor-Joining
Neighbor-Joining is the most commonly used method as it is the most computationally efficient of the four methods described. It is used for assessing large amounts of data with low divergence among sequences, much like Minimum Evolution. It also uses the distance matrix like Minimum Evolution, but also applies a cluster algorithm to construct the tree. This assigns sequences to particular groups and clusters them together based on those assignments.
Maximum Likelihood
Because of their complexity, Maximum Likelihood trees take a longer time to construct. However, it is a more effective and accurate tree because it uses a process that optimizes branch length by calculating tree scores. This method is also good at handling missing data.
Maximum Parsimony
Maximum Parsimony's main goal is to minimize the number of changes or nodes that occur in a tree. It produces accurate results in an efficient way. Parsimony is determined by assessment of the number of changes per site as well as the total character length for all species. There are three kinds of sites. The first, constant sites do not vary at all among sequences and therefor have a length of zero at all times among all trees. Singleton sites vary by only one and thus always have a length of one. Last, informative sites are sites with two different characteristics observed two or more times.
|
Discussion
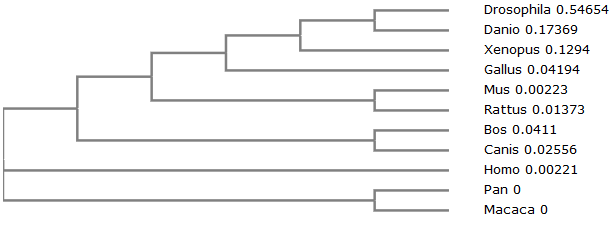
In this phylogenetic tree, we can see that the branch leading to human is not split from any other species besides the common ancestor. From this, we can determine that homo sapiens can be considered the outlier for this particular tree. However, this does not mean that we cannot use the homologs to study how MSTN works in humans. References
|
|
[1]"phylogeny". Biology online.
Figures
1. https://onaquasirelatednote.wordpress.com/2012/12/20/round-up-ready-best-of-2012-edition/all-the-birds-in-the-world/
2. http://www.cs.us.es/~fran/students/julian/_images/parts_of_a_phylogenectic_tree.png
2. http://www.cs.us.es/~fran/students/julian/_images/parts_of_a_phylogenectic_tree.png
This site was created as part of a project for Genetics 564
